Generalized Product Function Energy Partitioning
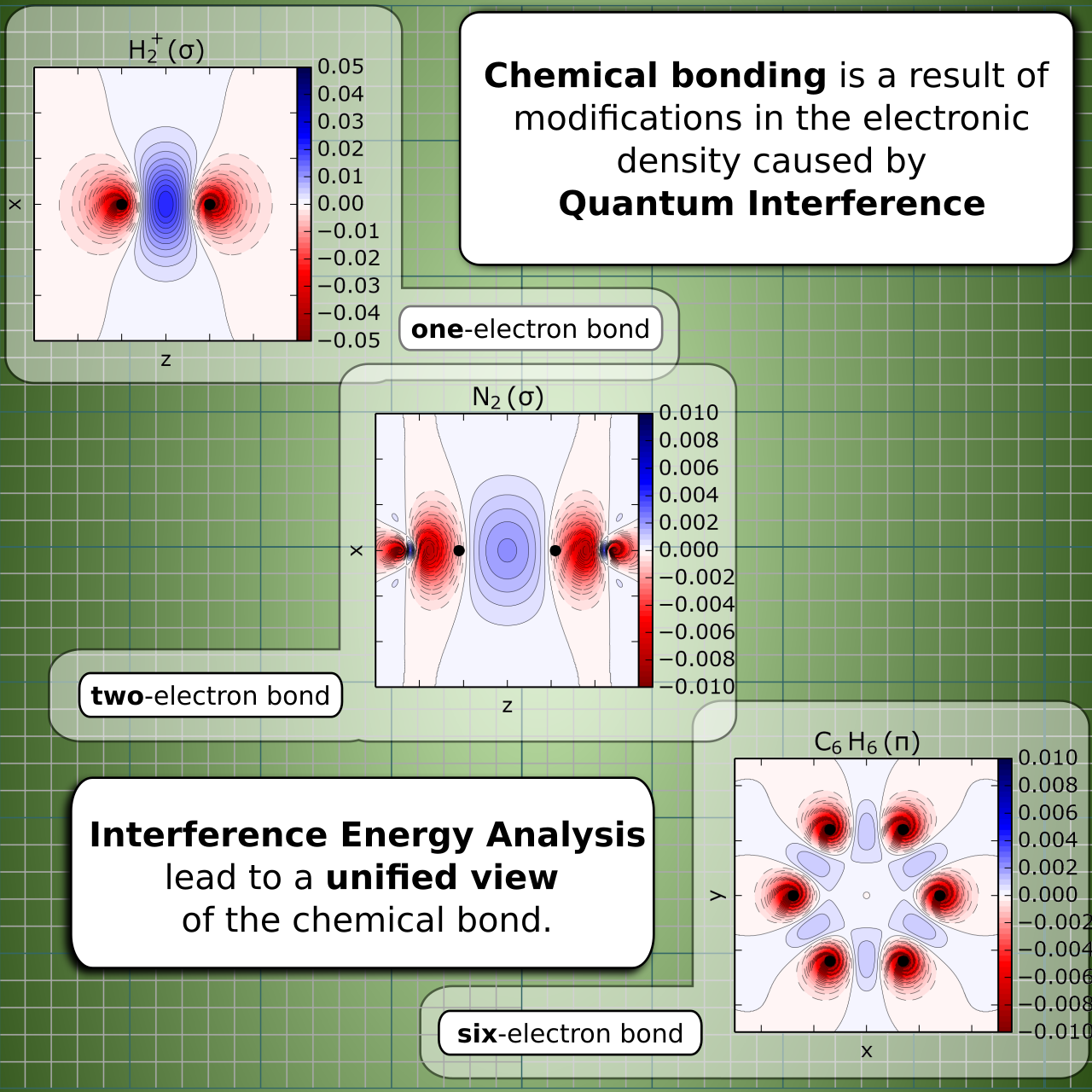
What is GPF-EP?
In simple words, the Generalized Product Function Energy Partitioning (GPF-EP) is a powerful Energy Decomposition Analysis (EDA) scheme suited for studying the nature of the chemical bond in molecular systems. The method is grounded on the original density-based energy decomposition first proposed by Ruedenberg [1]. Electronic densities are separated in a “quasi-classical” part, which can be mostly interpreted classically, and an interference part, which accounts for quantum interference between one-electron eigenstates. An analogous separation can be made for the electronic energy:

![E = E[QC] + E[INT]](img/energy_equation.svg)
GPF-EP is an adaptation of Rudenberg’s original framework to the GPF wave functions, developed by Roy McWeeny [2]. A GPF wave function consists of a strongly orthogonal product of smaller wave functions of electron subspaces (groups) of a molecular system, and its reduced density matrices (RDMs) consist of intergroup and intragroup parts. This particular structure of the RDMs of GPF wave functions allows the energy partitioning to be carried out for individual bonds and bond groups. The interpretation of the results is facilitated by the use of modern VB functions such as the Spin Coupled Generalized Valence Bond (SCGVB) [3,4], since that method naturally generate uniquely determined quasi-atomic orbitals, allowing direct correspondence between results and chemical structures (atoms connected by chemical bonds). This choice of group wave functions also precludes the selection of criteria for atomic orbital generation, which was somewhat arbitrary in Ruedenberg's original work.
About the Program
At the present version (1.0), the GPF-EP program exists as a subprogram of the GAMESS / VB2000 (3.0) code. It is planned to be fully integrated to VB2000 in the next version (3.1).
GPF-EP allows to partition the total energy of an atomic or molecular system into two main contributions, the quasi-classical energy, E[QC] and the interference energy, E[INT]. The GPF wave function allows the partition to be made for each electron group in a GPF wave function, which can be associated to properties of specific parts in a molecule (specific chemical bonds, lone pais, etc.).
There is also an implemented feature in the GPFEP program to partition the electric dipole moment of a molecule in the quasi-classical and interference contributions for each electron group (see reference 16 for further details).
Acknowledgements
The GPF-EP Authors (Thiago M. Cardozo, David W.O. de Sousa, Carlos E. V. Moura, and Marco A. C. Nascimento) are grateful to the Brazilian financial support agencies CAPES, CNPQ and FAPERJ.
The GPF-EP Program was originally written as part of TMC’s PhD thesis and further developed in DWOS’ MSc project. Besides the authors mentioned above, the following people have also contributed in this project: Gabriel Freitas Nascimento, Francisco Senna Vieira, and Felipe Fantuzzi.
GPFPlot
GPF-PLOT is a Python script which generates 2D plots of orbitals, electronic densities and GPF density partitioning associated to VB2000 output files. It is available for download on GitHub.
More Information
GPF-EP original paper: Cardozo, T. M.; Nascimento, M. A. C. Energy partitioning for generalized product functions: The interference contribution to the energy of generalized valence bond and spin coupled wave functions. J. Chem. Phys., 2009 130 (10) 104102. DOI: 10.1063/1.3085953
GPF-EP mini review: Fantuzzi, F.; Sousa, D. W. O. de; Nascimento, M. A. C. The Nature of the Chemical Bond from a Quantum Mechanical Interference Perspective. ChemistrySelect, 2017, 2 (2), 604–619. DOI: 10.1002/slct.201601535
GPF-EP Book Chapter and Tutorial: Cardozo, T. M.; Sousa, D. W. o. de; Fantuzzi, F.; Bitzer, R. S.; Nascimento, M. A. C. The Chemical Bond as a Manifestation of Quantum Mechanical Interference: Theory and Applications of the Interference Energy Analysis using SCGVB wave functions. In S. Shaik and P.C. Hiberty (eds.), Valence Bond, Elsevier, 2022. To be published.
GPF-EP Tutorial: Cardozo, T. M.; Sousa, D. W. o. de; Nascimento, M. A. C. The Quantum Interference Energy Analysis: A Tutorial. In S. Shaik and P.C. Hiberty (eds.), Valence Bond, Elsevier, 2022. To be published.
Also see a full list of the GPF-EP publications below.
References
Theory:
- Ruedenberg, K. The Physical Nature of the Chemical Bond. Rev. Mod. Phys. 1962, 34, 326–376. DOI: 10.1103/RevModPhys.34.326
- McWeeny, R. The density matrix in many-electron quantum mechanics I. Generalized product functions. Factorization and physical interpretation of the density matrices. Proc. R. Soc. A 1959, 253, 242–259. DOI: 10.1098/rspa.1959.0191
- Goddard, W. Improved Quantum Theory of Many-Electron Systems. I. Construction of Eigenfunctions of Ŝ² Which Satisfy Pauli's Principle. Phys. Rev. 1967, 157, 73–80; ibid., 81–93. DOI: 10.1103/PhysRev.157.73
- Gerratt, J.; Lipscomb, W. N. Spin-coupled wave functions for atoms and molecules.Proc. Natl. Acad. Sci. USA 1968, 59, 332–335. DOI: 10.1073/pnas.59.2.332
GPF-EP publications (updated Oct 16th, 2021):
- Cardozo, T. M.; Nascimento, M. A. C. Energy partitioning for generalized product functions: The interference contribution to the energy of generalized valence bond and spin coupled wave functions. J. Chem. Phys., 2009 130 (10) 104102. DOI: 10.1063/1.3085953
- Cardozo, T. M.; Nascimento, M. A. C. Chemical Bonding in the N2 Molecule and the Role of the Quantum Mechanical Interference Effect. J. Phys. Chem. A, 2009, 113 (45), 12541–12548. DOI: 10.1021/jp903963h
- Cardozo, T. M.; Nascimento Freitas, G.; Nascimento, M. A. C. Interference effect and the nature of the π-bonding in 1,3-butadiene. J. Phys. Chem. A, 2010, 114 (33), 8798–8805. DOI: 10.1021/jp101785p
- Fantuzzi, F.; Cardozo, T. M.; Nascimento, M. A. C. The role of quantum-mechanical interference and quasi-classical effects in conjugated hydrocarbons. Phys. Chem. Chem. Phys., 2012, 14 (16), 5479–88. DOI: 10.1039/c2cp24125k
- Vieira, F. S.; Fantuzzi, F.; Cardozo, T. M.; Nascimento, M. A. C. Interference energy in C-H and C-C bonds of saturated hydrocarbons: Dependence on the type of chain and relationship to bond dissociation energy. J. Phys. Chem. A, 2013, 117 (19), 4025–4034. DOI: 10.1021/jp4005746
- Cardozo, T. M.; Fantuzzi, F.; Nascimento, M. A. C. The non-covalent nature of the molecular structure of the benzene molecule. Phys. Chem. Chem. Phys., 2014, 16 (22), 11024. DOI: 10.1039/c3cp55256j
- Fantuzzi, F.; Nascimento, M. A. C. Description of polar chemical bonds from the quantum mechanical interference perspective. J. Chem. Theor. Comput., 2014, 10 (6), 2322–2332. DOI: 10.1021/ct500334f
- Fantuzzi, F.; Cardozo, T. M.; Nascimento, M. A. C. Nature of the Chemical Bond and Origin of the Inverted Dipole Moment in Boron Fluoride: A Generalized Valence Bond Approach. J. Phys. Chem. A, 2015, 119 (21), 5335–5343. DOI: 10.1021/jp510085r
- Sousa, D. W. O. de; Nascimento, M. A. C. Is There a Quadruple Bond in C2? J. Chem. Theor. Comput., 2016, 12 (5), 2234–2241. DOI: 10.1021/acs.jctc.6b00055
- Fantuzzi, F.; Cardozo, T. M.; Nascimento, M. A. C. The Nature of the Singlet and Triplet States of Cyclobutadiene as Revealed by Quantum Interference. ChemPhysChem, 2016, 17 (2), 288–295. DOI: 10.1002/cphc.201500885
- Fantuzzi, F.; Sousa, D. W. O. de; Nascimento, M. A. C. The Nature of the Chemical Bond from a Quantum Mechanical Interference Perspective. ChemistrySelect, 2017, 2 (2), 604–619. DOI: 10.1002/slct.201601535
- Fantuzzi, F.; Sousa, D. W. O. de; Nascimento, M. A. C. Chemical bonding in the pentagonal-pyramidal benzene dication and analogous isoelectronic hexa-coordinate species. Comput. Theor. Chem., 2017, 1116), 225–233. DOI: 10.1016/j.comptc.2017.03.020
- Fantuzzi, F.; Cardozo, T. M.; Nascimento, M. A. C. On the metastability of doubly charged homonuclear diatomics. Phys. Chem. Chem. Phys., 2017, 19 (29), 19352–19359. DOI: 10.1039/C7CP02792C
- Sousa, D. W. O. de; Nascimento, M. A. C. Are One-Electron Bonds Any Different from Standard Two-Electron Covalent Bonds? Acc. Chem. Res., 2017, 50 (9), 2264–2272. DOI: 10.1021/acs.accounts.7b00260
- Sousa, D. W. O. de; Nascimento, M. A. C. Quantum Interference Contribution to the Dipole Moment of Diatomic Molecules. J. Phys. Chem. A, 2018, 122 (5), 1406–1412. DOI: 10.1021/acs.jpca.7b11760
- Fantuzzi, F.; Wolff, W.; Quitián-Lara, H. M.; Boechat-Roberty, H. M.; Hilgers, G.; Rudek, B.; Nascimento, M. A. C. Unexpected reversal of stability in strained systems containing one-electron bonds. Phys. Chem. Chem. Phys., 2019, 21 (45), 24984–24992, 2019. DOI: 10.1039/C9CP04964A
- Sousa, D. W. O. de; Nascimento, M. A. C. One-electron bonds are not “half-bonds”. Phys. Chem. Chem. Phys., 2019, 21 (24), 13319–13336. DOI: 10.1039/C9CP02209K
- Sousa, D. W. O. de; Nascimento, M. A. C. Three-center two-electron bonds in the boranes B2H6 and B3H8− from the quantum interference perspective. Theor. Chem. Acc., 2020, 139 (8), 140. DOI: 10.1007/s00214-020-02654-4
- Sousa, D. W. O. de; Nascimento, M. A. C. Substituent Effects on the Quantum Interference of Two-Center One-Electron Bonds: [B 2X6]− (X = H, F, Cl, CN, OH, CH3, and OCH3). J. Phys. Chem. A, 2021, 125 (21), 4558–4564. DOI: 10.1021/acs.jpca.1c02771
- Sousa, D. W. O. de; Nascimento, M. A. C. Three-center two-electron bonding from the quantum interference perspective. To be published.